ФЯПЊПЦбаЭХЖгдкдЄВтЛљвђБэДяМАНтЪЭЗЧБрТыБфвьЗНУцЛёЈживЊНјеЙ
ЁЁЁЁФЯПЊаТЮХЭјбЖНќШеЃЌЮваЃШэМўбЇдКНВЪІдјЭёіЉЭХЖгдкдЄВтЛљвђБэДяМАНтЪЭЗЧБрТыБфвьЗНУцЛёживЊНјеЙЃЌбаОПГЩЙћвд“Reusabilityreport:Compressingregulatorynetworkstovectorsforinterpretinggeneexpressionandgeneticvariants”ЮЊЬтЃЌЗЂБэдкNatureзгПЏЁЖздШЛ•ЛњЦїжЧФмЁЗЃЈNatureMachineIntelligenceЃЉЩЯЃЌЦкПЏгАЯьвђзг15.508ЁЃ
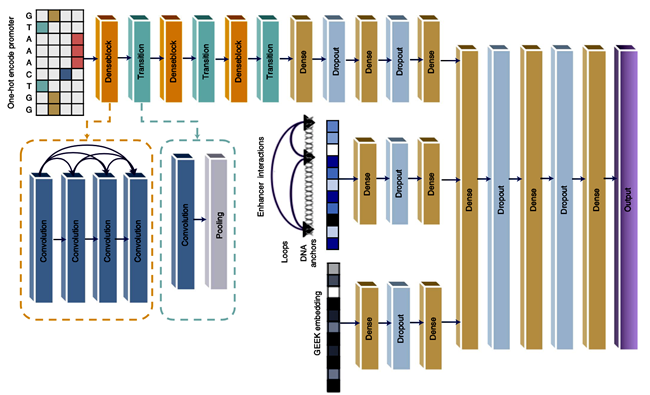
ЁЁЁЁШЋЛљвђзщЙиСЊбаОПЃЈGWASЃЉЪЧШЗЖЈИДдгМВВЁЗчЯеЛљвђЁќ/ЮЛІЦЕуЕФгааЇбаОПВпТдЃЌЮЊИДІЦдгМВВЁЕФбаОПжИУїСЫЗНЯђЃЌЮЊЪЕЯжИіадЛЏеяЖЯЁЂдЄКѓКЭжЮСЦЕьЖЈСЫМсЪЕЛљДЁЃЌДйНјСЫШЫРрвХДЋбЇКЭЛљвђЁозщбЇбаОПЕФЗЂеЙЁЃШЛЖјGWASбаОПЫљЕУЕФДѓЖрЪ§ЗчЯеЮЛЕуЖМЮЛгкЗЧБрТыЧјЃЌЩаВЛЧхГўетаЉЮЛЕуЪЧЗёжБНггыМВВЁЙІФмЯрЙиЁЃЖјОЋзМ eНтЪЭGWASЗЧБрТыЧјЕФЮЛЕуЪЧЪЕЯжОЋзМвНСЦЕФживЊЯШОіЁљЬѕМўЁЃ
ЁЁЁЁЕЅКЫмеЫсЖрЬЌадЃЈSNPsЃЉжївЊЪЧжИдкЛљвђзщЫЎЦНЩЯгЩЕЅИіКЫмеЫсЈLЕФБфвьЃЈМюЛљЕФзЊЛЛЛђЕпЛЛЁЂВхШыЛђШБЪЇЃЉЫљв§Ц№ЕФDNAађСаЖрЬЌадЁЃЫќЪЧШЫРрПЩвХДЋБфвьжазюГЃМћЕФЁавЛжжЁЃДѓЖрЪ§ЁЙЕФЙІФмадЗЧБрТыSNPsПЩвдЭЈЙ§ИЩЁЙШХзЊТМвђзгЕФНсКЯКЭЕїПидЊМўЕФЙІФмРДИФБфЛљвђЕФБэДяЃЌДгЖјЗЂЛгЦфзїгУЁЃжЕЕУзЂвтЕФЪЧЉIЁЁЃЌетаЉЕїПидЊМўОпгаИпЖШЕФЯИАћРраЭЬивьадЃЌетЬсЪОSNPsЕФЙІФмадвВгаЯИАћРраЭЬивьадЁЃвђДЫашвЊдке§ШЗЕФзщЁ§жЏКЭЧјгђБГОАЯТЖдУПвЛжжЯИАћРраЭжаЛюдОЕФЕїНкдЊМўНјааЗжРрКЭЙІФмНвЁїЪОЃЌНсКЯЦфЫљаЮГЩЕФЕїПиЭјТчЃЌНјЖјАяжњВћУїГЃМћЩёОЭЫааадМВВЁЗжзгЗЂВЁЛњжЦжаЕФЂйЛљвђЗчЯеЮЛЕуЙІФмЕФживЊадЁЃ
ЁЁЁЁНќФъРДЃЌЖрзщбЇММЪѕгШЦфЪЧБэЙлШОЩЋжЪзДЬЌКЭШ§ЮЌНсЙЙВтађММЪѕЕУЕНСЫЗЩЫйЗЂеЙгыЈЙуЗКгІгУЃЌетЖдНтЮіетаЉЗЧБрЁєТыЧјгђБфвьЕФЕїПиЛњжЦЁєДјРДСЫаТЕФЦѕЛњЁЃЮЊЯЕЭГНтЮіЕїПиЭјТчЖдЗЧБрТывХДЋБфвьЕФгАЯьЃЌбаОПЭХЖгЛљгкЭХЖгжЎЧАбаЈЗЂЕФDeepExpressionФЃаЭЃЌНјвЛВНећКЯађСаЪ§ОнЁЂHiChIPШ§ЮЌЛљвђзщЪ§ОнМАGEEKФЃаЭЕїПиЭјТчЪ§ОнЕФЕЭЮЌЯђЈСПБэЪОЃЈ(lowdimensionalemeddingЃЉЃЌЬсИпСЫЖдЛљвђБэДяЕФдЄВтФмСІЁЃЯТгЮбаОПНсЙћБэ eУїЃЌећКЯађСаЪ§ОнЁЂШ§ЮЌЛљвђзщЪ§ОнКЭЕїПиЭјТчЪ§ОнЃЌФмИќКУЕиРэНтзЊТМЕїПиЛњжЦЁЦЁЁКЭЗЧБрТыБфвьЕФЙІФмЃЌЮЊИќОЋзМНтЪЭGWASвХДЋБфвьЬсЙЉСЫаТЕФЗНЗЈЁЃ
ЁЁЁЁФЯПЊДѓбЇШэМўбЇЁюдКЮЊБОЮФЕквЛЕЅЮЛЃЌФЯПЊДѓбЇШэМўбЇдКНВЪІдјЭёіЉКЭЫЙЬЙИЃДѓбЇВЉЪПКѓаХЈОЇбЉЮЊЙВЭЌЕквЛзїепЃЌЧхЛЊДѓбЇГЄЦИИБНЬЪкНШ№КЭжаПЦдКЪ§бЇЫљЭѕгТбаОПдБЮЊТлЮФЕФЙВЭЌЭЈбЖзїепЁЃИУбаОПЁМвбЕУЕНЙњМвздШЛПЦбЇЛљН№ЧрФъЯюФПзЪжњЁЂЙњМвжиЕубаЗЂЧрФъПЦбЇМвЯюФПзЪжњЁЃЃЈШэІиЁЁМўбЇдКЙЉИхЃЉ
ЁЁЁЁТлЮФСДНгЃКhttps://www.nature.com/articles/s42256-021-00371-6
ЮДОдЪаэВЛЕУзЊдиЃКЖўОХЁПФъЛЊДѓбЇУХЛЇ » ФЯПЊПЦбаЭХЖгдкдЄВтЛљвђБэДяМАНтЪЭЗЧБрТыБфвьЗНУцЛёживЊНјеЙ
ЯрЙиЭЦМі
- ЁОЮвКЭФЯПЊЕФЙЪЪТЁПЮвгавЛЖЮЧщ
- ЁОзщЭМЁПНђФЯаЃЧјРњЪЗИДНЈЧјЃКЙсЭЈРњЪЗ аНЛ№ЯрДЋ
- НЬг§ЁќВПЕГЪЗбЇЯАНЬг§ИпаЃЕкЖўжИЕМзщВЮМгЮваЃАьЙЋЪвЁЂБЃУмАьЕГжЇВПЕквЛЕГаЁзщзЈЬтзщжЏЩњЛюЛс
- ЮваЃВЮМгШЋЙњНЬг§ЯЕЭГвпЧщЗРПиЙЄзїЪгЦЕЕїЖШЛсвщ
- ФЯПЊбЇзгдкЕкЈЪЎвЛНьЖргяжжШЋЙњПквыДѓШќжаЛёМбМЈ
- ЮКДѓХєТЪЬьНђЪаеўаЮЏдБДњБэЭХЗУЮЪЮваЃ
- ФЯПЊДѓбЇбаОПЭХЖгдкЭиЦЫЙтзгбЇСьЈ}гђдйЛёаТНјеЙ
- ФЯПЊбЇзгдкЁАаТЪБДњ аТЧрФъЬИИФИяПЊЗХЁБИпаЃУНЬхЭјЦРзїЦЗеїМЏЛюЖЏжаЛёМбМЈ
- КТбЧУїЃКжжзхЦчЪггыГ№ЭтжївхбЯжизшАШЋЈшЧђвпЧщЗРПи
- ЗЂЛгХХЧђг§ШЫЙІФм МљааФЯПЊЬхг§ОЋЩё
- ЙњЮёдКгІЖдаТЙкЗЮбзвпЧщСЊЗРСЊПиЛњжЦзлКЯзщжТаХИаЁэаЛФЯПЊбЇеп
- ФЯПЊДѓбЇейПЊ2020ФъЖШаЃСьЕМАрзгКЭСьЕМИЩВППМКЫМАИЩВПбЁАЮШЮгУЁАвЛБЈИцСНЦРвщЁБЙЄзїЛс
- ФЯПЊДѓбЇ2021аТФъвєРжЛсдкЬьНђДѓЁђОчдКОЋВЪЩЯбн
- КЃФЯЪЁНЬг§ЬќЬќГЄВмЯзРЄвЛааРДЗУЮваЃ
- ФЯПЊДѓбЇНЬЪІдкЕкЦпНьШЋЙњЕчЙЄЕчзгЛљДЁПЮЁшГЬЪЕбщНЬбЇАИР§ЩшМЦОКШќжаЛёМбМЈ
- ЕчЪгОчЁЖЮвУЧЕФЮїФЯСЊДѓЁЗзпНјФЯПЊ
- ФЯПЊДѓбЇЗжзгЮЂЩњЮябЇгыММЪѕНЬг§ВПжиЕуЪЕбщЪв2020ФъЖШбЇЪѕЮЏдБЛсейПЊ
- ЁОгАйЁЉФъаЃЧьЁППЦММШеБЈЩчзмБрМСѕбЧЖЋзіПЭЁААйФъФЯПЊДѓНВЬГЁБ
- ЁОгАйФъаЃЧьЁПФЯОЉДѓбЇеХвьБіНЬЪкЈзіПЭЁААйФъФЯПЊДѓНВЬГЁБ
- ФЯПЊДѓбЇейПЊЕГЮЏГЃЮЏЃЈРЉДѓЃЉЛсєпжааФзщбЇЯАЛс
аТЮХЙЋИц
- ФЯПЊжњФуЁАДДЁБЧрДК 12-08
- бюЧьЩНМьВщбЇаЃАВШЋЙЄзї 12-07
ИпПМеаЩњ
- ФЯПЊДѓбЇ2018ФъЈеаЩњеТГЬ 08-05
бЇаЃРЯЪІ
- Тоаћ
- УчдТФў
- ЧисА
- ЮКНЁмА
- ЛЦезЛљ
- МжКщВЈ
- СѕжиСІ
- РюгЈ
- ГЃЯўОВ
- ЙШРіЮА
- ЭѕЯш
- ЫеЙ№еф
- РюжОИе
- РюЯцКН
- АВРћ
- ЦыКщЭњ
- аЯФНаТ
- еХЪїжк
- ЭѕКъИе
- НЊИЃбѓ
- ЦюУљ
- НЊЦМ
- ШюДѓЧП
- ЫїН№УЗ
- ГЄемРЩ
- ТэгЈ
- бюОВ
- ЬЦТЗбя
- бюЪРЧП
- ЗыГчвх
- ЙљУї
- ГТЯрЮФ
- ГТЕТЕк
- ЧњшЊ
- КЋБѓ
- вЖЯМ
- ЫяСЋЙѓ
- еХгёЬЋ
- ЭѕНЈгЂ
- ЙигЂ
- ХЃНЈСЂ
- ТэвхЦН
- РюОАгъ
- ЭѕНЈал
- ЭѕЯВХє
- СѕБѓ
- едгёКь
- Рюву
- АНв
- РюЧПЃЈЩњПЦЃЉ